
Compartmentalized redox and metabolic remodeling in a tumor microenvironment. 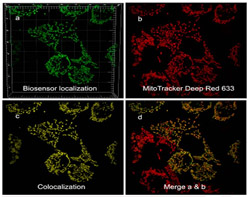
Cells expressing a malignant phenotype generally exhibit altered energy metabolism, resistance to apoptosis and increased production of reactive oxygen species, all of which reflect changes in mitochondrial metabolism. Compartmentalized redox homeostasis is a key determinant in the extent to which these alterations lead to the emergence of genetic instability and increasingly autonomous and aggressive behavior.
However, compartmentalized redox homeostasis is a difficult area of study and little is known regarding the dynamics of glutathione potentials among distinct compartments. It is also now clear that tumor invasiveness and malignant progression is strongly affected by the tumor microenvironment, which is also a difficult phenomenon to study.
We are using genetically encoded GFP-based redox biosensors and microfluidic 3D cell culture platforms to: (a) test hypothesis that RAS-driven glutaminolysis in transformed cancer cells reflects in part increased use of glutamine-derived carbon for cytosolic synthesis of the most abundant redox buffer in eukaryotic cells, glutathione; (b) determine the preferred metabolic pathways fueling the migratory/malignant phenotype of glioma stem cells in glioblastoma.
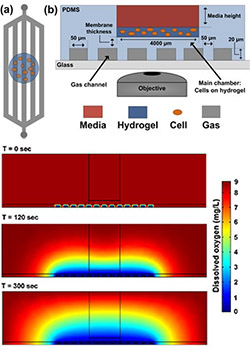
Figure 1 (right): Simplified schematic of the PDMS-based microfluidic platform for microenvironmental studies. (a) top-down and (b) cross-section views. Cells are introduced in bolus into the cell chamber along with media, experimental reagents, and redox calibrants. c) Computational model simulating the amount of dissolved oxygen within the microfluidic platform at different time points after introducing introducing an anoxic gas stream.
Mitochondria-to-nucleus retrograde signaling by redox-sensitive CHCHD2 in glioblastoma
Glioblastoma (GBM) is the most common and malignant form of brain cancer in adults, with a median survival time of 12-15 months. GBM tumors present with chemo- and radio-resistance and diffusive invasiveness, making them impossible to completely resect and resulting in high rates of recurrence. Although amplification and mutation of the epidermal growth factor receptor (EGFR) occurs in 50% of GBM cases, treatment modalities targeting this receptor have remained largely ineffective, suggesting the presence of adaptive compensatory signaling networks conferring such resistance. Amplification of chromosome 7p11.2, which includes the EGFR locus, is frequently associated with the expression of a constitutively active, ligand-independent mutant form of the receptor termed EGFRvIII. Tumor expression of EGFRvIII is correlated with shorter median survival and is present in 50-60% of patients whose tumors display genetic amplification of EGFR.
Proximal to EGFR on Chr. 7p11.2 is a gene encoding the mitochondrial protein coiled-coil-helix-coiled-coil-helix domain-containing protein 2 (CHCHD2). CHCHD2 contains a C-terminal CHCH domain consisting of two CX9C motifs, each characterized by two thiol containing redox-sensitive cysteines separated by 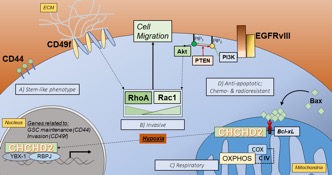 9 amino acids. First described as a protein necessary for optimal oxidative phosphorylation, CHCHD2 has been further implicated in signaling between mitochondria and the nucleus. This phenomenon, termed direct mitochondria-to-nucleus retrograde signaling, allows mitochondria and cells to reach homeostasis following fluctuations in internal conditions or stress. This inter-organelle communication has the potential to play a crucial role in cancer cells, particularly in GBM, which readily adapt to microenvironmental stresses and chemotherapeutics. Our recent work provides evidence that CHCHD2 translocation from mitochondria to nucleus is magnified in the presence of EGFRvIII in U87 GBM cells and in response to hypoxia, both of which drive stemness and malignancy in GBM. Accordingly, we are testing the overall hypothesis that the intracellular redox environment of hypoxic cells favors the accumulation of nuclear CHCHD2, which in turn plays a role in mediating or driving stemness in GBM, and that this capability is enhanced in GBM cells harboring the EGFRvIII mutation. An understanding of the nature, triggers, and consequences of this compensatory retrograde signaling will identify dynamic pathways that can be leveraged and targeted in treating GBM.
9 amino acids. First described as a protein necessary for optimal oxidative phosphorylation, CHCHD2 has been further implicated in signaling between mitochondria and the nucleus. This phenomenon, termed direct mitochondria-to-nucleus retrograde signaling, allows mitochondria and cells to reach homeostasis following fluctuations in internal conditions or stress. This inter-organelle communication has the potential to play a crucial role in cancer cells, particularly in GBM, which readily adapt to microenvironmental stresses and chemotherapeutics. Our recent work provides evidence that CHCHD2 translocation from mitochondria to nucleus is magnified in the presence of EGFRvIII in U87 GBM cells and in response to hypoxia, both of which drive stemness and malignancy in GBM. Accordingly, we are testing the overall hypothesis that the intracellular redox environment of hypoxic cells favors the accumulation of nuclear CHCHD2, which in turn plays a role in mediating or driving stemness in GBM, and that this capability is enhanced in GBM cells harboring the EGFRvIII mutation. An understanding of the nature, triggers, and consequences of this compensatory retrograde signaling will identify dynamic pathways that can be leveraged and targeted in treating GBM.
Taurocholic acid metabolism by gut microbes and colon cancer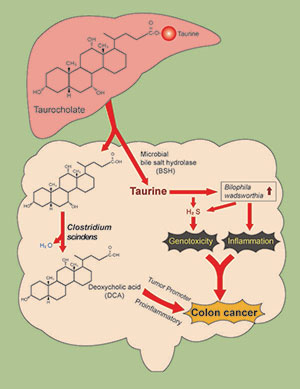
We are examining a novel mechanism that might explain why consumption of a high red meat and saturated fat diet imparts risk for colorectal cancer (CRC) development involving primary microbial risk factors (bile acid metabolizing and sulfidogenic bacteria) that are modifiable by diet. The focus is on the biliary metabolite taurocholic acid (3α,7α,12α-trihydroxy-5β-cholan-24-oic acid N-(2-sulfoethyl)amide) (TCA). Gut microbes in the ileum and colon deconjugate TCA to cholic acid (CA) liberating taurine, which is used by the colonic bacterium Bilophila wadsworthia as a substrate for anaerobic respiration generating genotoxic hydrogen sulfide (H2S). In addition, CA is further metabolized by colonic bacteria to the proinflammatory and tumor promoting secondary bile acid deoxycholate (DCA). Thus, the components of TCA, taurine and CA, support the growth of microbial groups who, through interaction of their metabolic by-products, are implicated mechanistically in DNA-damage and tumor-promotion. The secondary bile acid DCA acts as a tumor promoter by causing membrane perturbations leading to the release of arachidonic acid, which is converted by the enzymes COX-2 and lipooxygenase, to pro-inflammatory and pro-angiogenic prostaglandins, and reactive oxygen species which damage DNA and inhibit DNA repair enzymes.
Our published data highlight the role of H2S as a bacterial-derived intestinal insult that leads to activation of proinflammatory pathways and genomic instability—two hallmarks of CRC. Specifically, we have demonstrated that: (a) H2S induces proliferative, proinflammatory and DNA repair pathways in nontransformed intestinal epithelial cells, (b) exogenous H2S is a potent genotoxin at concentrations many-fold less than those measured in the human colon, (c) H2S induces free radical-based DNA damage in the absence of cellular metabolism,19 and (d) colonic mucosa of healthy human subjects is persistently colonized by sulfidogenic bacteria which utilize both inorganic and organic sources of sulfur including the taurine respiring bacterium B. wadsworthia.